Crystal Generation¶
The very first step of ASSYST is the generation of random, periodic crystal structures.
Imports¶
from assyst.crystals import Formulas, sample_space_groups
from assyst.plot import concentration_histogram, distance_histogram
Formulas¶
The basic unit for structure generation in ASSYST is a formula unit of a given material, such as:
Mg$_2$Ca
Mg$_4$Ca$_2$
and so on
This specifies the number of atoms in a target structure and ASSYST tries to generate as many different space groups of each of these as possible.
Creation¶
They can by manually constructed
mg = Formulas(
({'Mg': 1}, {'Mg': 2}, {'Mg': 3}, {'Mg': 4})
)
mgca = Formulas(
({'Mg': 2, 'Ca': 1}, {'Mg': 4, 'Ca': 2})
)
or with some helper methods
Formulas.range('Mg', 1, 5)
Formulas(atoms=({'Mg': 1}, {'Mg': 2}, {'Mg': 3}, {'Mg': 4}))
Operations¶
Formulas.range('Mg', 1, 5) | Formulas.range('Ca', 1, 3)
Formulas(atoms=({'Mg': 1, 'Ca': 1}, {'Mg': 2, 'Ca': 2}))
Formulas.range('Mg', 1, 5) * Formulas.range('Ca', 1, 3)
Formulas(atoms=({'Mg': 1, 'Ca': 1}, {'Mg': 1, 'Ca': 2}, {'Mg': 2, 'Ca': 1}, {'Mg': 2, 'Ca': 2}, {'Mg': 3, 'Ca': 1}, {'Mg': 3, 'Ca': 2}, {'Mg': 4, 'Ca': 1}, {'Mg': 4, 'Ca': 2}))
Formulas.range('Mg', 1, 5) + Formulas.range('Ca', 1, 3)
Formulas(atoms=({'Mg': 1}, {'Mg': 2}, {'Mg': 3}, {'Mg': 4}, {'Ca': 1}, {'Ca': 2}))
Sampling¶
Once the formulas are defined the structures can be generated.
atoms = sample_space_groups(
formulas = mgca,
)
The sampler returns a generator, the actual structures can be obtained by collecting it in a list.
atoms = list(atoms)
Visualization¶
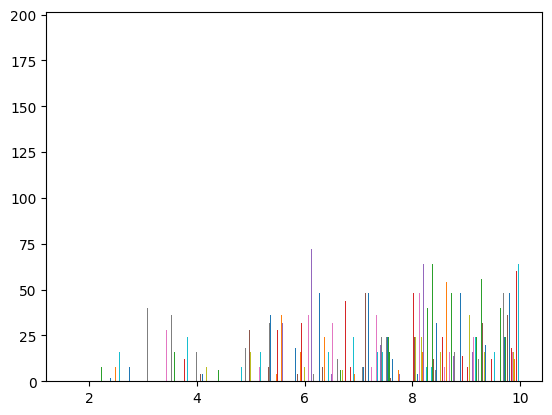
distance_histogram(atoms, rmax=10, reduce=lambda x: x);
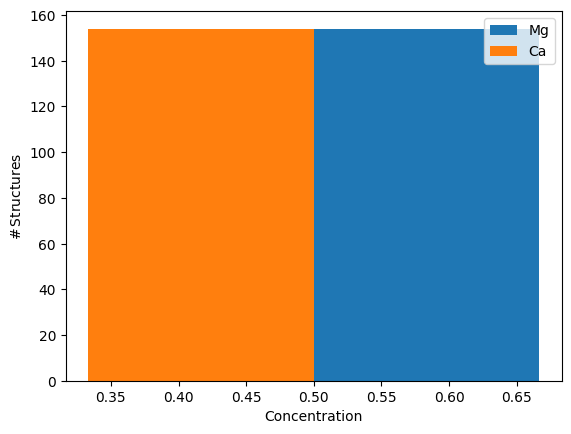
concentration_histogram(atoms)
Advanced Sampling¶
The sampling function supports a few more options for advanced sampling.
sample_space_groups?
Signature:
sample_space_groups(
formulas: Union[assyst.crystals.Formulas, Iterable[dict[str, int]]],
spacegroups: Union[list[int], tuple[int, ...], Iterable[int], NoneType] = None,
min_atoms: int = 1,
max_atoms: int = 10,
max_structures: int | None = None,
dim: Literal[0, 1, 2, 3] = 3,
tolerance: Union[Literal['metallic', 'atomic', 'molecular', 'vdW'], assyst.filters.DistanceFilter, dict] = 'metallic',
) -> Iterator[ase.atoms.Atoms]
Docstring:
Create symmetric random structures.
Args:
formulas (Formulas or iterable of dicts from str to int): list of chemical formulas
spacegroups (list of int): which space groups to generate
max_atoms (int): do not generate structures larger than this
max_structures (int): generate at most this many structures
dim (one of 0, 1, 2, or 3): the dimensionality of the structures to generate; if lower than 3 the code generates
samples no longer from space groups, but from the subperiodic layer, rod, or point groups.
tolerance (str, dict of elements to radii):
specifies minimum allowed distances between atoms in generated structures;
if str then it should be one values understood by :class:`pyxtal.tolerace.Tol_matrix`;
if dict each value gives the minimum *radius* allowed for an atom, whether a given distance is allowed then
depends on the sum of the radii of the respective elements
Yields:
`Atoms`: random symmetric crystal structures
File: ~/science/phd/dev/assyst/assyst/crystals.py
Type: function