A Complete Workflow for a Single Element¶
This notebook runs the full ASSYST pipeline end-to-end on a unary system (copper). The four canonical steps are:
Sampling — generate symmetric random structures for every space group.
Relaxing — minimise volume first, then full cell + positions, to find the relevant pockets of the potential energy surface.
Perturbing — apply random rattles and stretches around those minima to populate the surrounding PES.
Combining — collect everything, filter unphysical configurations, evaluate the reference and store the resulting training data.
In a real workflow the relaxations and final evaluations are carried out with DFT (or any other reference); here we use a Morse potential so the notebook runs quickly.
Imports¶
import pickle
from pathlib import Path
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
from tqdm.auto import tqdm
from assyst.crystals import Formulas, sample
from assyst.filters import DistanceFilter, AspectFilter, VolumeFilter
from assyst.relaxations import VolumeRelax, FullRelax, relax
from assyst.perturbations import RandomChoice, Rattle, Stretch, perturb
Configuration¶
max_num is the largest number of atoms per generated structure. 2 keeps
the example fast; 10 is the typical production value and yields ~10k
training structures.
max_num = 2
out_dir = Path(f"Unary/{max_num}")
out_dir.mkdir(parents=True, exist_ok=True)
Reference potential¶
Any ASE calculator works. We pick a Morse potential whose parameters were chosen so that its minimum sits near the experimental Cu-Cu bond length.
For more interesting reference data the universal Grace graph-ACE models
in assyst.calculators are a good drop-in replacement (they require the
optional tensorpotential dependency).
from ase.calculators.morse import MorsePotential
reference = MorsePotential(epsilon=0.3, r0=2.55265548 * 1.10619396, rho0=4)
1. Sampling random structures¶
Build the formulas to sample (Cu, Cu_2, …, up to max_num) and let
sample generate one structure for every compatible space group.
AspectFilter(6) filters out very elongated unit cells that pyxtal
occasionally produces.
formulas = Formulas.range('Cu', max_num + 1)
formulas
Formulas(atoms=({'Cu': 0}, {'Cu': 1}, {'Cu': 2}))
spg = list(filter(
AspectFilter(6),
sample(
formulas,
spacegroups=range(1, 230 + 1),
max_atoms=max_num,
)
))
print(f"Generated {len(spg)} symmetric seeds")
Generated 167 symmetric seeds
Each entry is an :class:ase.Atoms carrying the sampled cell and positions. A small summary of the first few sampled structures:
pd.DataFrame([
{
'formula': s.get_chemical_formula(),
'natoms': len(s),
'volume_per_atom': s.cell.volume / len(s),
}
for s in spg[:5]
])
| formula | natoms | volume_per_atom | |
|---|---|---|---|
| 0 | Cu | 1 | 10.911529 |
| 1 | Cu | 1 | 11.704551 |
| 2 | Cu | 1 | 11.899922 |
| 3 | Cu | 1 | 12.247109 |
| 4 | Cu | 1 | 11.184826 |
Just to create a small check point, dump generated structures into a pickle file. This is not required for assyst.
with open(out_dir / 'spg.pckl', 'wb') as f:
pickle.dump(spg, f)
2. Relaxation¶
ASSYST applies a two-step relaxation:
VolumeRelax— relax the volume only, keeping shape and internal positions frozen. Quickly puts each seed near a sensible energy scale.FullRelax— relax cell shape, volume and atomic positions together, landing in a (local) minimum of the PES.
In production both would be carried out with DFT; here the Morse potential
makes the cost negligible. For experimentation, assyst.relaxations also
provides CellRelax and SymmetryRelax.
volset = VolumeRelax(max_steps=100, force_tolerance=1e-3)
allset = FullRelax(max_steps=100, force_tolerance=1e-3)
%%time
volmin = list(relax(tqdm(spg, desc='volume'), volset, reference))
CPU times: user 1min 2s, sys: 55.4 ms, total: 1min 2s
Wall time: 21 s
%%time
allmin = list(relax(tqdm(volmin, desc='full'), allset, reference))
CPU times: user 4min, sys: 414 ms, total: 4min
Wall time: 1min 25s
relax returns a new list of :class:ase.Atoms. Comparing the volume per atom before and after each relaxation step shows how the cells shrink towards their minima:
pd.DataFrame({
'sampled': [s.cell.volume / len(s) for s in spg[:5]],
'volmin': [s.cell.volume / len(s) for s in volmin[:5]],
'allmin': [s.cell.volume / len(s) for s in allmin[:5]],
})
| sampled | volmin | allmin | |
|---|---|---|---|
| 0 | 10.911529 | 15.667167 | 11.761170 |
| 1 | 11.704551 | 13.639392 | 11.761452 |
| 2 | 11.899922 | 44.776388 | 12.829227 |
| 3 | 12.247109 | 13.261506 | 12.829213 |
| 4 | 11.184826 | 54.634555 | 298.602200 |
with open(out_dir / 'volmin.pckl', 'wb') as f:
pickle.dump(volmin, f)
with open(out_dir / 'allmin.pckl', 'wb') as f:
pickle.dump(allmin, f)
3. Random perturbations¶
Around each minimum we sample the surrounding PES with three families of random perturbations described in the ASSYST paper:
mostly positional noise with a small cell change (
rattle),mostly hydrostatic cell change,
mostly shear cell change.
The hydrostatic and shear strains are combined with RandomChoice so that
hydrostatic changes are drawn slightly more often. The final mods
sequence is applied to every fully-minimised structure. DistanceFilter
discards perturbations that placed atoms unphysically close.
rattle = Rattle(0.25) + Stretch(hydro=0.05, shear=0.005)
hydro = Stretch(hydro=0.80, shear=0.05)
shear = Stretch(hydro=0.05, shear=0.20)
stretch = RandomChoice(hydro, shear, 0.7)
# four rattles + four stretches per minimum reproduces the published settings
mods = 4 * [rattle] + 4 * [stretch]
%%time
random = list(perturb(allmin, mods, filters=[DistanceFilter({'Cu': 1})]))
print(f"Produced {len(random)} perturbed structures")
Produced 1188 perturbed structures
CPU times: user 497 ms, sys: 6.98 ms, total: 504 ms
Wall time: 429 ms
Each relaxed minimum is expanded into a cloud of perturbed structures. Looking at the spread of volumes per atom around the first minimum gives a feel for the noise applied:
ref = allmin[-1]
v0 = ref.cell.volume / len(ref)
siblings = [s for s in random if s.info['seed'] == ref.info['seed']]
pd.DataFrame([
{
'natoms': len(s),
'volume_per_atom': s.cell.volume / len(s),
'rel_volume_change': s.cell.volume / len(s) / v0 - 1,
}
for s in siblings
])
| natoms | volume_per_atom | rel_volume_change | |
|---|---|---|---|
| 0 | 2 | 11.764033 | 0.000484 |
| 1 | 2 | 11.468011 | -0.024692 |
| 2 | 2 | 11.943824 | 0.015774 |
| 3 | 2 | 10.899526 | -0.073039 |
| 4 | 2 | 11.717989 | -0.003432 |
| 5 | 2 | 11.878374 | 0.010208 |
| 6 | 2 | 11.415293 | -0.029175 |
| 7 | 2 | 11.321541 | -0.037148 |
with open(out_dir / 'random.pckl', 'wb') as f:
pickle.dump(random, f)
4. Combine and evaluate¶
Concatenate the sampled, relaxed and perturbed structures, run the distance and volume filters once more to reject any leftover pathological configurations, and finally evaluate the reference to obtain a training dataset of energies and forces.
everything = list(filter(
VolumeFilter(300),
filter(DistanceFilter({'Cu': 1}), spg + volmin + allmin + random),
))
print(f"Final training set: {len(everything)} structures")
Final training set: 1565 structures
rows = []
for s in tqdm(everything, desc='evaluating'):
s.calc = reference
rows.append({
'ase_atoms': s,
'energy': s.get_potential_energy(),
'forces': s.get_forces(),
'number_of_atoms': len(s),
})
df = pd.DataFrame(rows)
df.to_pickle(out_dir / 'everything_training_data.pckl.gz')
df.head()
| ase_atoms | energy | forces | number_of_atoms | |
|---|---|---|---|---|
| 0 | (Atom('Cu', [-1.0386276484191481, -0.194573218... | -2.168733 | [[3.191891195797325e-16, -2.8622937353617317e-... | 1 |
| 1 | (Atom('Cu', [0.6736784829368637, -1.1197747671... | -2.354713 | [[-7.592633932598097e-17, 6.227072826023132e-1... | 1 |
| 2 | (Atom('Cu', [0.0, -1.1901120180848723, -7.2873... | -1.787319 | [[4.0722633598555547e-16, -8.066464163292153e-... | 1 |
| 3 | (Atom('Cu', [0.0, 0.0, -1.3521411060002535], i... | -1.345679 | [[-2.0643209364124004e-16, -5.204170427930421e... | 1 |
| 4 | (Atom('Cu', [-1.0311953554480588, -1.031195355... | -1.358783 | [[-3.226585665316861e-16, -2.42861286636753e-1... | 1 |
Inspect the training data¶
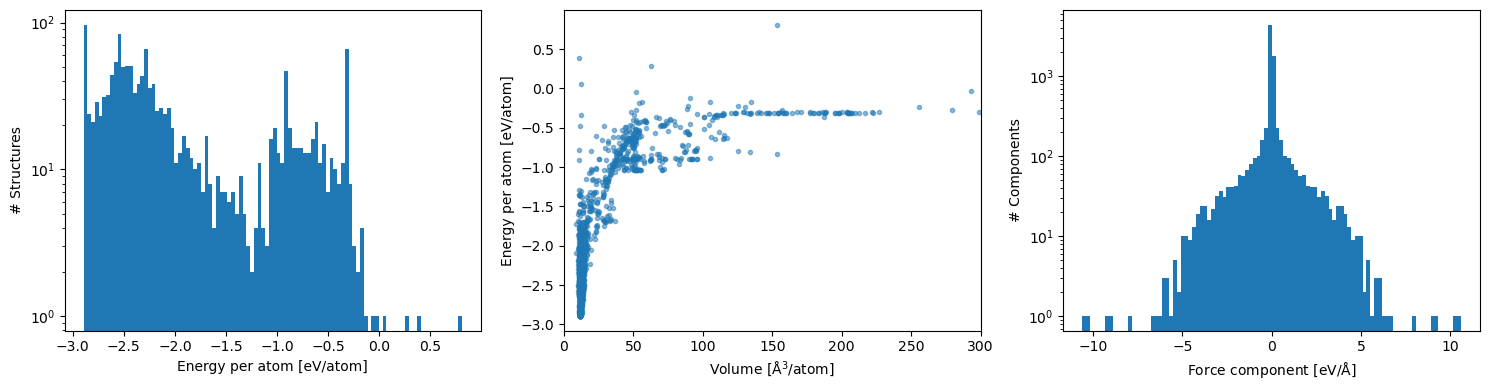
Energy per atom, energy vs volume and force component distributions are a quick sanity check that the dataset spans the expected ranges.
fig, axes = plt.subplots(1, 3, figsize=(15, 4))
(df['energy'] / df['number_of_atoms']).plot.hist(bins=100, log=True, ax=axes[0])
axes[0].set_xlabel('Energy per atom [eV/atom]')
axes[0].set_ylabel('# Structures')
axes[1].scatter(
df['ase_atoms'].map(lambda s: s.cell.volume / len(s)),
df['energy'] / df['number_of_atoms'],
marker='.', alpha=0.5,
)
axes[1].set_xlim(0, 300)
axes[1].set_xlabel(r'Volume [$\mathrm{\AA}^3$/atom]')
axes[1].set_ylabel('Energy per atom [eV/atom]')
df['forces'].explode().explode().infer_objects().plot.hist(bins=100, log=True, ax=axes[2])
axes[2].set_xlabel(r'Force component [eV/$\mathrm{\AA}$]')
axes[2].set_ylabel('# Components')
plt.tight_layout()
Optional: post-hoc filtering¶
If the resulting energies and forces still cover ranges that are too
extreme, EnergyFilter and ForceFilter (or pandas query) can prune
them. These act on ase.Atoms objects that carry a
SinglePointCalculator, which is what relax and the loop above
attach.
df.query('-3 <= energy / number_of_atoms < 10')
| ase_atoms | energy | forces | number_of_atoms | |
|---|---|---|---|---|
| 0 | (Atom('Cu', [-1.0386276484191481, -0.194573218... | -2.168733 | [[3.191891195797325e-16, -2.8622937353617317e-... | 1 |
| 1 | (Atom('Cu', [0.6736784829368637, -1.1197747671... | -2.354713 | [[-7.592633932598097e-17, 6.227072826023132e-1... | 1 |
| 2 | (Atom('Cu', [0.0, -1.1901120180848723, -7.2873... | -1.787319 | [[4.0722633598555547e-16, -8.066464163292153e-... | 1 |
| 3 | (Atom('Cu', [0.0, 0.0, -1.3521411060002535], i... | -1.345679 | [[-2.0643209364124004e-16, -5.204170427930421e... | 1 |
| 4 | (Atom('Cu', [-1.0311953554480588, -1.031195355... | -1.358783 | [[-3.226585665316861e-16, -2.42861286636753e-1... | 1 |
| ... | ... | ... | ... | ... |
| 1560 | (Atom('Cu', [0.1381167850060396, -0.1182780644... | -4.905344 | [[-3.971783227794896, 1.8061865367255856, 1.07... | 2 |
| 1561 | (Atom('Cu', [0.0, 0.0, 0.0], index=0), Atom('C... | -4.404489 | [[-1.345494896054511e-15, -7.971054372113429e-... | 2 |
| 1562 | (Atom('Cu', [0.0, 0.0, 0.0], index=0), Atom('C... | -4.565983 | [[-5.88938620094126e-16, -6.934557095217286e-1... | 2 |
| 1563 | (Atom('Cu', [0.0, 0.0, 0.0], index=0), Atom('C... | -4.377709 | [[-4.077282877100984e-15, 5.11729872886002e-16... | 2 |
| 1564 | (Atom('Cu', [0.0, 0.0, 0.0], index=0), Atom('C... | -5.381921 | [[3.2067447655603765e-15, 2.912600716165059e-1... | 2 |
1565 rows × 4 columns
Next steps¶
This notebook stops at the training set. Fitting a potential is then the
job of a downstream MLIP package — e.g. ACE via python-ace, MTP via
mlip-3, or any other framework that reads a list of structures with
energies and forces. The dataframe saved in
Unary/<max_num>/everything_training_data.pckl.gz is in the format
consumed by pyace.linearacefit.LinearACEDataset.
The companion notebook ACEFitting.ipynb walks through training a small linear ACE potential on the dataset produced here.